Cu ce se confundă scleroza laterală amiotrofică? Scleroza laterala amiotrofica
Scleroza laterală amiotrofică sau boala Lou Gehrig este o boală rapid progresivă a sistemului nervos caracterizată prin afectarea neuroni motorii măduva spinării, cortexul și trunchiul cerebral. De asemenea, în proces patologic sunt implicate ramurile motorii ale neuronilor cranieni (trigemen, facial, glosofaringian).
Epidemiologia bolii
Boala este extrem de rară, aproximativ 2-5 persoane la 100.000 Se crede că bărbații de peste 50 de ani sunt mai des afectați. Boala lui Lou Gehrig nu face excepții pentru nimeni, afectează oameni de toate dimensiunile. statut socialși diverse profesii (actori, senatori, laureați Premiul Nobel, ingineri, profesori). Cel mai faimos pacient a fost campionul mondial de baseball Loi Gering, după care boala și-a luat numele.
În Rusia, scleroza laterală amiotrofică a devenit larg răspândită. În prezent, numărul persoanelor bolnave este de aproximativ 15.000-20.000 în populație. Printre oameni faimosi Rusia, având această patologie, putem aminti compozitorul Dmitri Șostakovici, politicianul Yuri Gladkov și cântărețul pop Vladimir Migulya.
Cauzele sclerozei laterale amiotrofice
Boala se bazează pe acumularea de proteine patologice insolubile în celulele motorii ale sistemului nervos, ducând la moartea acestora. Cauza bolii este momentan necunoscută, dar există multe teorii. Principalele teorii includ:
- Viral - această teorie a fost populară în anii 60-70 ai secolului al XX-lea, dar nu a fost niciodată confirmată. oameni de știință din SUA iar URSS a efectuat experimente pe maimuțe, injectându-le cu extracte din măduva spinării persoanelor bolnave. Alți cercetători au încercat să demonstreze participarea la formarea bolii.
- Ereditar - în 10% din cazuri patologia este ereditară;
- Autoimună - această teorie se bazează pe descoperirea unor anticorpi specifici care ucid celulele nervoase motorii. Există studii care demonstrează formarea unor astfel de anticorpi pe fundalul altora boală severă(de exemplu, când cancer de plamani sau limfom Hodgkin);
- Genetică - 20% dintre pacienți au tulburări ale genelor care codifică enzima foarte importantă Superoxid dismutaza-1, care convertește substanțele toxice celule nervoase Superoxid la oxigen;
- Neuronal - Oamenii de știință britanici cred că elementele gliale, adică celulele care asigură activitatea vitală a neuronilor, sunt implicate în dezvoltarea bolii. Cercetările au arătat că atunci când astrocitele, care elimină glutamatul din terminații nervoase, probabilitatea de a dezvolta boala Lou Gehrig crește de zece ori.
Clasificarea sclerozei laterale amiotrofice:
Simptomele sclerozei laterale amiotrofice
Orice formă de boală are același debut: pacienții se plâng de creștere slabiciune musculara, scădea masa muscularași apariția fasciculațiilor (smușcări musculare).
Forma bulbară a SLA caracterizat prin simptome de afectare a nervilor cranieni (9, 10 și 12 perechi):
- Cei bolnavi au vorbirea și pronunția deteriorate și devine dificil să-și miște limba.
- De-a lungul timpului, actul de înghițire este întrerupt, pacientul se sufocă în mod constant, iar mâncarea poate curge prin nas.
- Pacienții simt o zvâcnire involuntară a limbii.
- Progresia SLA este însoțită de atrofia completă a mușchilor feței și gâtului, pacienții le lipsesc complet expresiile faciale, nu pot deschide gura sau mesteca alimente.
Varianta cervicotoracică Boala afectează în primul rând membrele superioare ale pacientului, simetric pe ambele părți:
- Inițial, pacienții simt o deteriorare a funcționalității mâinilor, devine mai greu să scrieți, să jucați instrumente muzicale, efectuați mișcări complexe.
- În același timp, mușchii brațului sunt foarte tensionați și reflexele tendinoase sunt crescute.
- În timp, slăbiciunea se extinde la mușchii antebrațului și ai umărului, se atrofiază. Membrului superior seamănă cu un bici atârnat.
Forma lombo-sacrala de obicei începe cu o senzație de slăbiciune la nivelul extremităților inferioare.
- Pacienții se plâng că le-a devenit mai dificil să lucreze în picioare, să meargă pe distanțe lungi și să urce scările.
- În timp, piciorul începe să se lase, mușchii picioarelor se atrofiază, iar pacienții nici măcar nu pot sta în picioare.
- Apar reflexe tendinoase patologice (Babinsky). Pacienții dezvoltă incontinență urinară și fecală.
Indiferent de varianta care predomină la pacienții la începutul bolii, rezultatul este în continuare același. Boala progresează constant, răspândindu-se la toți mușchii corpului, inclusiv la mușchii respiratori. Când mușchii respiratori eșuează, pacientul începe să aibă nevoie ventilatie artificiala plămâni și îngrijire constantă.
În practica mea, am observat doi pacienți cu SLA, un bărbat și o femeie. Se distingeau prin culoarea părului roșu și vârsta relativ fragedă (până la 40 de ani). În exterior, erau foarte asemănătoare: nu avea nicio urmă de mușchi, o față amiabilă și gura lor era întotdeauna ușor deschisă.
Astfel de pacienți mor în majoritatea cazurilor din cauza boli concomitente(pneumonie, sepsis). Chiar și cu îngrijirea corespunzătoare, dezvoltă escare (vezi), pneumonie ipostatică. Dându-și seama de severitatea bolii lor, pacienții cad în depresie, apatie și încetează să fie interesați de lumea de afarași pe cei dragi tăi.
În timp, psihicul pacientului suferă schimbări puternice. Un pacient pe care l-am văzut timp de un an era capricios, labilitate emoțională, agresivitate, lipsă de reținere. Testele de inteligență au arătat o scădere a gândirii sale, abilități mentale, memorie, atenție.
Diagnosticul sclerozei laterale amiotrofice
Principalele metode de diagnosticare includ:
- RMN-ul măduvei spinării și al creierului– metoda este destul de informativă evidențiază atrofia părților motorii ale creierului și degenerarea structurilor piramidale;
- puncție cefalorahidiană– de obicei dezvăluie normal sau continut crescut veveriţă;
- examene neurofiziologice– electroneurografie (ENG), electromiografie (EMG) și stimulare magnetică transcraniană (TCMS).
- analiza genetică moleculară– studii ale genei care codifică Superoxid dismutaza-1;
- test biochimic de sânge– detectează o creștere de 5-10 ori a creatin fosfokinazei (o enzimă formată în timpul descompunerii musculare), crestere usoara enzime hepatice (ALT, AST), acumularea de deșeuri în sânge (uree, creatinina).
Ce se întâmplă în ALS
Datorită faptului că SLA are simptome similare cu alte boli, se pune diagnosticul diferențial:
- boli ale creierului: tumori ale fosei posterioare, atrofie a sistemelor multiple,
- boli musculare: miodistrofie oculofaringiană, miotonie Rossolimo-Steinert-Kurshman
- boli sistemice
- boli ale măduvei spinării: leucemie sau limfom limfocitar, tumori ale măduvei spinării, amiotrofie spinală, siringomielie etc.
- boli nervi periferici: Sindromul Personaj-Turner, neuromiotonia lui Isaac, neuropatie motorie multifocală
- miastenia gravis, sindromul Lambert-Eaton - boli ale joncțiunii neuromusculare
Tratamentul sclerozei laterale amiotrofice
Tratamentul bolii este în prezent ineficient. Medicamente iar îngrijirea adecvată a pacientului nu face decât să prelungească speranța de viață fără a asigura o recuperare completă. Terapie simptomatică include:
- Riluzol (Rilutek)– un medicament bine dovedit în SUA și Marea Britanie. Mecanismul său de acțiune este de a bloca glutamatul din creier, îmbunătățind astfel funcționarea Superoxid Dismutaza-1.
- interferența ARN este o metodă foarte promițătoare de tratare a SLA, ai cărei creatori au primit Premiul Nobel pentru Medicină. Tehnica se bazează pe blocarea sintezei proteinelor patologice în celulele nervoase și prevenirea morții ulterioare a acestora.
- Transplant de celule stem– studiile au arătat că transplantul de celule stem în central sistem nervos previne moartea celulelor nervoase, restabilește conexiuni neuronale, îmbunătățește creșterea fibrelor nervoase.
- Relaxante musculare – elimină spasmele musculare și spasmele (Baclofen, Sirdalud).
- Anabolice (Retabolil)– pentru a crește masa musculară.
- Medicamente anticolinesterazice(Prozerin, Kalimin, Pyridostigmine) – previne distrugerea rapidă a acetilcolinei în sinapsele neuromusculare.
- vitaminele B(Neurorubin, Neurovitan), vitaminele A, E, C - aceste medicamente îmbunătățesc conducerea impulsurilor de-a lungul fibrelor nervoase.
- Antibiotice gamă largă actiuni(cefalosporine din generația 3-4, fluorochinolone, carbopeneme) – indicat pentru dezvoltare complicatii infectioase, septicemie.
ÎN terapie complexă includ neapărat hrănirea printr-o sondă nazogastrică, masaj, terapie cu exerciții fizice cu un medic și consultații cu un psiholog.
Prognoza
Din păcate, prognosticul pentru scleroza laterală amiotrofică este nefavorabil. Pacienții mor literalmente în câteva luni sau ani, durata medie viata la pacienti:
- doar 7% trăiesc mai mult de 5 ani
- cu debut bulbar - 3-5 ani
- pentru lombar - 2,5 ani
Un prognostic mai favorabil pentru cazurile ereditare ale bolii asociate cu mutații ale genei superoxid dismutază-1.
Situația din Rusia este încețoșată de faptul că pacienților nu li se acordă îngrijire adecvată, dovadă fiind faptul că Riluzote, un medicament care încetinește progresia bolii, nici măcar nu a fost înregistrat în Rusia până în 2011 și doar în același timp. anul, boala în sine a fost inclusă în lista „rară”. Dar la Moscova există:
- Fond pentru ajutarea pacienților cu reacții adverse scleroza amiotrofică la Centrul Mercy Marfo-Mariinsky
- Fundația caritabilă G.N.Levitsky pentru pacienții cu SLA
În cele din urmă, aș dori să adaug despre evenimentul caritabil Ice Bucket Challenge care a avut loc în iulie 2014. A avut ca scop strângerea de fonduri pentru sprijinirea pacienților cu scleroză laterală amiotrofică și a devenit destul de răspândită. Organizatorii au reușit să strângă peste 40 de milioane de dolari.
Esența acțiunii a fost că o persoană fie s-a stropit cu o găleată apa cu gheatași îl surprinde pe video sau donează o anumită sumă de bani unei organizații caritabile. Evenimentul a devenit destul de popular datorită participării unor interpreți populari, actori și chiar politicieni.
Încetinește progresia bolii și prelungește perioada de boală în care pacientul nu are nevoie de îngrijire externă constantă.
Reduceți severitatea simptomelor individuale ale bolii și mențineți un nivel stabil al calității vieții.
Indicații pentru spitalizare
Examenul primar.
Efectuarea gastrostomiei endoscopice percutanate.
Aspecte etice și deontologice ale managementului pacienților cu scleroză laterală amiotrofică
Diagnosticul de SLA poate fi comunicat pacientului numai după o examinare amănunțită, care nu este întotdeauna o examinare unică. Uneori este necesar un EMG repetat. Conform Convenției Helsinki de Bioetică (1997), medicii trebuie să informeze pacienții cu boli incurabile despre un diagnostic care necesită decizii legate de moartea iminentă. Diagnosticul de SLA trebuie comunicat într-o manieră sensibilă, subliniind variabilitatea progresiei bolii. Sunt cunoscute cazuri de progresie extrem de lentă (cu purtător homozigot al mutației D90A) și în cazuri sporadice izolate. Trebuie amintit că 7% dintre pacienți trăiesc mai mult de 60 de luni. Neurologul trebuie să stabilească un contact strâns cu pacientul și familia acestuia și să comunice diagnosticul în prezența familiei și a prietenilor, într-un mediu calm, confortabil pentru pacient, fără grabă. La întrebările pacientului trebuie să se răspundă cu așteptarea reacției sale emoționale. Nu poți spune unui pacient că nu se poate face nimic pentru a-l ajuta. Dimpotrivă, ar trebui să-l convingi să consulte un neurolog sau centru specializat la fiecare 3-6 luni Este necesar să subliniem că simptomele individuale răspund bine la tratament.
Terapie medicamentoasă
Terapia patogenetică
Singurul medicament care încetinește semnificativ progresia SLA este riluzolul, un inhibitor presinaptic al eliberării glutamatului. Utilizarea medicamentului permite prelungirea vieții pacienților cu o medie de 3 luni. Riluzolul este indicat pacienților cu SLA certă sau probabilă, după excluderea altora cauze probabile leziuni ale neuronilor motori periferici și centrali, cu o durată a bolii mai mică de 5 ani, accelerate capacitate vitala plămâni (FVC) mai mult de 60%, fără traheostomie. Pentru pacienții cu posibil SLA cu o durată mai mică de 5 ani, FVC<60% и трахеостомией для предотвращения аспирации без зависимости от аппарата ИВЛ рилузол, согласно мнению экспертов, также может быть показан. Препарат назначают в дозе 100 мг в день вне связи с приёмом пищи. Каждые 3 мес необходимо мониторировать уровень АЛТ, АСТ и ЛДГ из-за риска развития лекарственного гепатита. Концентрация рилузола в сыворотке крови несколько ниже у мужчин и курильщиков, поэтому рекомендуется уменьшить количество выкуриваемых сигарет или прекратить курение. Рилузол следует принимать пожизненно.
S-au încercat terapia patogenetică a SLA cu alte medicamente, dar toate acestea, inclusiv factori neutrofici, xaliproden (un ligand cu molecularitate scăzută al receptorilor factorilor neurotrofici), anticonvulsivante (lamotrigină, gabapentin, topiramat etc.), agenți metabolici ( gangliozide, aminoacizi ramificati, creatina), medicamente antiparkinsoniene (selegilina), antibiotice (ciclosporina), antioxidanti (acetilcisteina, vitamina E), blocante ale canalelor de calciu (nimodipina, verapamil), imunomodulatorii (interferon beta, imunoglobulina) si altele au fost ineficiente.
Nu există date convingătoare privind eficacitatea dozelor mari de Cerebrolysin, deși utilizarea sa a condus la o activare generală a pacienților.
Îngrijire paliativă
Metodele de corectare a principalelor simptome ale ALS sunt prezentate în tabel. 34-5.
Tabelul 34-5. Îngrijiri paliative pentru SLA
| Simptom/indicație | Metode de corectare |
| Fasciculații, crampe | Carbamazepină 100 mg de două ori pe zi, baclofen 10-20 mg pe zi sau tizanidină cu o creștere treptată a dozei la 8 mg pe zi |
| Spasticitate | Baclofen 10-20 mg pe zi sau tizanidină cu o creștere treptată a dozei la 8 mg/zi, diazepam în doză de 2,5-5 mg de 3 ori pe zi |
| Depresie, labilitate emoțională | Amitriptilină până la 100 mg/zi noaptea, fluoxetină 20 mg/zi noaptea |
| Îmbunătățirea metabolismului muscular | Carnitină 250 mg, trei capsule de patru ori pe zi. Creatină 3 g/zi pentru varianta piramidală, 6 g/zi pentru varianta clasică și 9 g/zi pentru varianta segmento-nucleară a SLA. Soluție de levocarnitină 20%, 15 ml de 4 ori pe zi. Curs de terapie timp de două luni de trei ori pe an. Soluție de propionat de trimetilhidraziniu 10%, 10 ml la 200 ml de soluție de clorură de sodiu 0,9% intravenos (curs - 10 perfuzii, de 1-2 ori pe an) |
| Terapia cu multivitamine | Acid tioctic 600 mg pe zi timp de 2 săptămâni de 1-2 ori pe an. Multivitamine (milgamma 2 ml intramuscular zilnic timp de 2 săptămâni de 1-2 ori pe an, neuromultivit 2 capsule de 3 ori pe zi timp de 2 luni de 2 ori pe an) |
| Pareze peroneală, deformare a piciorului echinovar | Pantofi ortopedici |
| Pareza extensoare a gâtului | Suport de cap semirigid sau rigid |
| Tulburări de mers | Bastoane, plimbări, cărucioare |
| Oboseală | Amantadină 100 mg/zi timp de o lună, dacă este ineficientă - etosuximidă 37,5 mg/zi, dacă este ineficientă - gimnastică de 2 ori pe zi timp de 1 5 minute (exerciții cu contracție pasivă) |
| Tromboza venoasă profundă a extremităților inferioare | Înfășurare elastică pentru picioare |
| Contractura spastica a mainii | Atele relaxante |
| Periartroza humeroscapulară | Comprese cu dimetil sulfoxid 30% (o linguriță), procaină 0,25% (două lingurițe), 3 ml hialuronidază (dizolvați 64 de unități de pulbere) timp de 30-40 de minute timp de 3-5 zile |
| Salivaţie | Igienizarea mecanică sau medicinală a cavității bucale (clătire frecventă cu soluții antiseptice, periaj pe dinți de trei ori pe zi). Limitarea produselor lactate fermentate. Amitriptilină până la 100 mg/zi noaptea. Atropină 0,1% 1 ml, două picături în fiecare colț al gurii cu 10-20 minute înainte de masă și noaptea. Utilizarea sistemică a atropinei este plină de efecte secundare (tahicardie, constipație) |
| Sindromul de hipersecreție orală | Unități de aspirație portabile. Bronhodilatatoare și mucolitice (acetilcisteină 600 mg oral pe zi). Corectarea deshidratării |
| Disartrie | Relaxante musculare (vezi. «
spasticitate"). Aplicații de gheață pe limbă. Ghidurile de vorbire ale Asociației Britanice ALS. Mașini de scris electronice. Etran tabele cu litere sau cuvinte. Sistem informatic pentru tastarea caracterelor folosind senzori tactili instalați pe globii oculari |
| Disfagie | Mâncăruri piure și măcinate, piureuri, sufleuri, jeleuri, terci, agenți de îngroșare lichizi. Excluderea vaselor cu componente lichide și solide contrastante ca densitate. Gastrostomie endoscopică percutanată |
| Sindromul de apnee obstructivă în somn | Fluoxetină 20 mg/zi noaptea |
| Tulburări respiratorii (FVC)<60-70%) | Ventilație intermitentă neinvazivă |
O îmbunătățire a stării emoționale conform scalei calității vieții pentru SLA ALSAQ-40 și, în consecință, activarea generală a pacienților a fost detectată atunci când pacienții au fost tratați cu o soluție 1% de Semax (metionil-glutamil-histidil-fenilalanil-). prolil-glicil-prolină) intranazal în doză de 12 mg/zi (două cure de 10 zile cu pauză de 2 săptămâni). Acest medicament din grupul nootropelor nu are niciun efect asupra progresiei bolii.
Medicamentele miotrope metabolice care pot fi prescrise pentru SLA includ capsulele de carnitină, levocarnitina (soluție orală) sau propionat de trimetilhidraziniu (picurare intravenoasă), precum și creatina, în funcție de varianta bolii. Cu toate acestea, un studiu clinic recent al creatinei nu a confirmat efectul său pozitiv asupra scăderii forței musculare identificate în studiul original_ În varianta segmentar-nucleară a SLA cu debut lombar, apare mioliză pronunțată și nivelul seric al CPK crește, prin urmare se crede că utilizarea preparatelor de carnitină în astfel de cazuri este mai sigură de -datorită riscului de a dezvolta insuficiență renală acută în timpul tratamentului cu creatină. Dacă există o scădere semnificativă a activității motorii, medicamentele miotrope sunt întrerupte, deoarece în caz contrar vor crește catabolismul muscular. Din același motiv, nu se recomandă prescrierea nandrolonului, care, pe lângă efectul negativ indicat, duce la dezvoltarea impotenței. De asemenea, pentru ALS, se obișnuiește să se prescrie preparate multivitamine sau combinații de vitamine B cu preparate cu acid tioctic. Orice efect pozitiv al preparatelor miotrope și vitaminice asupra evoluției bolii nu a fost stabilit.
Un complex de tulburări motorii la pacienții cu boală neuronului motor necesită utilizarea metodelor de corecție ortopedică. În plus, în centrele specializate din străinătate există seturi de veselă și alte aparate de uz casnic convenabile pacienților. Pacienților ar trebui să li se explice că utilizarea acestor ajutoare nu „lipește” » etichetarea lor „cu dizabilități”, dar, dimpotrivă, le permite să reducă dificultățile asociate bolii, să mențină pacienții în cercul vieții sociale și, de asemenea, să îmbunătățească calitatea vieții rudelor și prietenilor lor.
S-a demonstrat că folosirea gimnasticii timp de 15 minute de două ori pe zi încetinește scăderea forței musculare și ajută la corectarea oboselii periferice în SLA. Unii autori au în vedere și metodele medicinale utilizate pentru scleroza multiplă, care fac posibilă corectarea oboselii de origine centrală în SLA (vezi Tabelul 34-5).
Una dintre cele mai importante domenii ale îngrijirii paliative este tratamentul tulburărilor bulbare și pseudobulbare. Ele apar la debutul bolii cu paralizie bulbară progresivă (forma bulbară a SLA) și se asociază în 67% din cazuri cu debutul coloanei vertebrale a SLA.
Producția de salivă în SLA este mai mică în comparație cu indivizii sănătoși. În același timp, pe măsură ce disfagia progresează, se dezvoltă saliva din cauza incapacității de a înghiți și de a scuipa excesul de saliva. Tratamentul paliativ al salivarii este important deoarece acest simptom contribuie la dezvoltarea infecțiilor oportuniste în cavitatea bucală, care la rândul lor crește manifestările de disfagie și disartrie, crește riscul de a dezvolta pneumonie de aspirație și, în final, creează disconfort emoțional și crește depresia, deoarece imaginea unei persoane cu salivă care curge din gură este asociată în rândul oamenilor obișnuiți cu demență, de care nu suferă pacienții cu boală neuronului motor.
Pe lângă amitriptilină (vezi Tabelul 34-5), metodele de combatere a salivației includ utilizarea de aspirație portabilă, injecții subcutanate de toxină botulină în doză de până la 120 de unități per glandă parotidă și până la 20 de unități per glandă submandibulară, iradierea de glandele salivare parotide, aplicarea de fluorouracil pe glandele salivare, timpanotomie. Toate aceste tratamente sunt considerate a fi inferioare ca eficacitate față de terapia cu amitriptilină, deși nu au fost efectuate studii clinice comparative. Salivația este doar o componentă a unui astfel de simptom precum hipersecreția orală, care este cauzată de igienizarea afectată a arborelui traheobronșic. Corectarea deshidratării la pacienții cu disfagie și deficiență nutrițională se realizează folosind perfuzii de glucoză 5%, dar nu de clorură de sodiu, pentru a preveni mielinoliza pontină centrală, manifestată prin sindrom vestibular acut în prezența unor tulburări bulbare preexistente.
Cel mai precoce simptom al acestui grup este disartria. Poate fi spastică și însoțită de nazofonie în variantele clasice și piramidale de SLA, sau flasc și însoțită de răgușeală în varianta nucleară segmentară. Disartria, spre deosebire de disfagie, nu este un simptom care pune viața în pericol, dar înrăutățește semnificativ calitatea vieții pacientului și îi limitează capacitatea de a participa la viața socială. Disartria în SLA cu prezența tetraparezei profunde înrăutățește semnificativ calitatea vieții persoanelor care îngrijesc pacientul, din cauza dificultăților care apar în acest caz în înțelegerea reciprocă între pacient și rudă. Cu toate acestea, disartria este cel mai dificil de tratat.
Disfagia este un simptom fatal al bolii neuronului motor, deoarece duce la dezvoltarea deficienței nutriționale (cașexie), imunodeficienței secundare, care crește simultan riscul de a dezvolta pneumonie de aspirație și infecții oportuniste. În stadiile inițiale, se efectuează igienizarea frecventă a cavității bucale, iar ulterior consistența alimentelor este schimbată.
Pacientului trebuie să i se explice că alimentele trebuie luate întotdeauna stând stând cu capul drept pentru a asigura cel mai fiziologic act de înghițire și pentru a preveni dezvoltarea pneumoniei de aspirație. Încă din primele etape ale disfagiei, pacientul este discutat cu pacientul despre necesitatea efectuării gastrostomiei endoscopice percutanate. S-a demonstrat că îmbunătățește starea pacienților cu SLA și le prelungește viața.
Această operație este indicată pentru o scădere a greutății corporale cu mai mult de 2% pe lună în prezența disfagiei; o încetinire pronunțată a actului de înghițire (mâncând un castron de terci mai mult de 20 de minute); restricție pronunțată a aportului de lichide cu amenințarea deshidratării (mai puțin de 1 litru de lichid pe zi); prezența leșinului hipoglicemic; FVC >50%.
O contraindicație pentru gastrostomia endoscopică este scăderea FVC<50%, поскольку во время операции при раздувании желудка возможна острая дыхательная недостаточность из-за воздействия на диафрагму и плевру с включением пульмонокардиального рефлекса. Перед операцией необходимо провести исследование трофического статуса пациента и назначить пероральную искусственную питательную смесь, чтобы предотвратить нарушения заживления послеоперационной раны на фоне иммунодефицита, а также антибиотики. к сожалению, пациенты с БАС редко соглашаются на проведение гастростомии в силу эмоциональных проблем, обусловленных невозможностью принимать пищу через рот. После операции проводят энтеральное питание искусственными питательными смесями в зависимости от трофического статуса и питательных потребностей больного, а также жидкими пищевыми продуктами (бульон, кисель в объёме до 400 мл) . При отказе от гастростомии проводят периодическое зондовое кормление искусственными питательными смесями с повышенным содержанием углеводов, парентеральное и ректальное питание. Назначаются эубиотики и пробиотики, растительные слабительные и большое количество жидкости.
Principalul simptom fatal al SLA este insuficiența respiratorie, care apare ca urmare a parezei și atrofiei diafragmei și a mușchilor respiratori accesorii sau a degenerarii centrului respirator al medulei oblongate. În primul rând, se alătură paraliziei bulbare progresive, cu debut difuz și toracic al SLA. În acest din urmă caz, ele apar mai repede decât la debutul cervical, datorită leziunii inițiale a mușchilor respiratori auxiliari și apoi principali. Odată cu apariția ALS cervicală, slăbiciunea mușchilor respiratori principali, de regulă, este compensată pentru o lungă perioadă de timp de funcția mușchilor auxiliari. Pacienții cu ALS dezvoltă insuficiență respiratorie restrictivă asociată cu o scădere a suprafeței ventilate a plămânilor, care ulterior se transformă în restrictiv-obstructiv din cauza unei încălcări a trecerii secrețiilor traheobronșice. Odată cu debutul bulbar al SLA, apare situația opusă, când insuficiența respiratorie obstructivă devine mixtă datorită adăugării unei componente restrictive.
Semnele timpurii ale problemelor de respirație includ simptome precum vise vii, amețeală dimineață, nemulțumire față de somn și somnolență în timpul zilei. Pentru depistarea precoce a tulburărilor respiratorii se efectuează spirografie și polisomnografie. Dacă este prezentă apneea în somn, se prescrie fluoxetină 20 mg noaptea timp de 3 luni. Pe viitor, se recomandă utilizarea dispozitivelor periodice de ventilație neinvazivă (BiPAP). Din păcate, aceste dispozitive sunt scumpe și, prin urmare, inaccesibile. Durata ședințelor variază de la 2 ore pentru tulburările ușoare până la 20 de ore, inclusiv noaptea, pentru cele severe. Se efectuează fluometria de vârf, determinarea gazelor din sânge și terapia cu oxigen. S-a demonstrat că ventilația neinvazivă a plămânilor, a început înainte de căderea FVC<60%, может продлить жизнь при БАС на 1 год. Гипербарическая оксигенация не эффективна. При потребности во вспомогательном дыхании свыше 20 ч ставят вопрос о переходе на инвазивную ИВЛ.
Nevoia de traheostomie și ventilație mecanică este un semnal de apropiere a morții. Argumentele împotriva efectuării ventilației mecanice pentru boala neuronului motor includ probabilitatea ca pacientul să fie scos din dispozitiv, dificultățile tehnice și costul ridicat al îngrijirii unui pacient dependent de un dispozitiv de ventilație mecanică, dezvoltarea tulburărilor extramotorii la pacienții care primesc ventilație mecanică. (demență, tulburări cerebeloase, extrapiramidale, senzoriale, pelvine), precum și complicații post-resuscitare (encefalopatie posthipoxică, pneumonie, tromboză venoasă profundă a extremităților inferioare, escare). În Statele Unite, costul îngrijirii unui pacient ventilat la domiciliu este de 200.000 USD pe an. Argumentele pentru efectuarea ventilației mecanice sunt dorința pacienților individuali de a-și prelungi viața, precum și cazurile rare de conservare a funcțiilor cognitive și chiar performanța parțială la un număr de pacienți cu SLA după transferul acestora la ventilație mecanică. În Japonia, 80% dintre pacienții cu SLA sunt transferați la ventilație mecanică, în SUA - 10%, în Marea Britanie - 1%. În nicio țară din lume ventilația nu este inclusă în asigurarea de sănătate, aceasta este asigurată doar pe cheltuiala familiei pacientului la domiciliu sau într-un cadru de îngrijire medicală. În plus, transferul la ventilația mecanică pentru SLA se efectuează numai dacă pacientul, în prezența unui avocat și a unui reprezentant legal, a convenit asupra condițiilor de deconectare de la dispozitiv.
Indicațiile clinice pentru trecerea la ventilație mecanică sunt sindromul bulbar izolat cu tulburări respiratorii sau insuficiența respiratorie spinală izolată cu tetrapareză, dar fără tulburări bulbare. În prezența tetraparezei și a tulburărilor bulbare, i.e. „sindrom blocat”, transferul la ventilație mecanică nu este indicat. Un transfer de urgență la ventilația mecanică, dacă este imposibil să obțineți instrucțiuni de la pacient cu privire la tactici ulterioare, nu se efectuează.
Terapie non-medicamentală
Nu există recomandări specifice pentru regimul pentru SLA. Se crede că activitatea fizică excesivă, inclusiv sportul, nu este indicată, deoarece un astfel de stil de viață înainte de dezvoltarea bolii este asociat cu riscul dezvoltării acesteia. Alimentele trebuie să fie suficiente, bogate în calorii, blânde și variate din punct de vedere mecanic și termic.
MANAGEMENTUL SUPLIMENTAR AL PACIENTULUI
După examenul final inițial sau repetat, care stabilește diagnosticul de SLA, pacienții trebuie să rămână sub observație ambulatorie (o dată la 3-6 luni) și să li se ofere consiliere în etape pe măsură ce apar noi simptome. Tratamentul cu medicamente metabolice miotrope și terapia cu vitamine se efectuează în cursuri, alte medicamente sunt luate continuu. Se recomandă efectuarea spirografiei la 3 luni și, dacă pacientul ia riluzol, după 3 luni, iar apoi la 6 luni pentru a determina activitatea ALT, AST și LDH. În prezența disfagiei și a deficienței nutriționale, trebuie evaluate starea trofică și nivelul glicemiei. Pentru a efectua gastrostomia endoscopică percutanată, pacientul este internat pentru scurt timp în spital, unde după operație este selectat volumul și frecvența optime de nutriție enterală. În cazul în care pacientul refuză această operație, el poate fi internat pentru o perioadă scurtă de timp pentru a fi supus terapiei prin perfuzie pentru a corecta deshidratarea sau hrănirea periodică cu tub. Daca pacientul nu dispune de ventilatia mecanica neinvaziva periodica si traheostomia cu transfer la ventilatie mecanica nu poate fi efectuata din motive legale sau medicale, este indicata oxigenoterapia. Dacă oxigenoterapia în volum de 2-4 l/min nu elimină dificultățile de respirație în repaus în decubit sau în șezut, este indicată prescrierea de analgezice narcotice (morfină în doză de 5 mg/zi în comprimate, sau în sub formă de supozitor rectal, sau subcutanat 1 ml de soluție 0,1% în doză de 25 mg/zi în tablete sau lorazepam în doză de 2 mg/zi în tablete; soluție orală sau supozitor rectal). Pacientul poate fi acasă sau plasat în hospice.
PROGNOZA
Prognosticul pentru SLA este întotdeauna nefavorabil, cu excepția cazurilor ereditare rare asociate cu anumite mutații ale genei superoxid dismutază -1 (D90A și altele). Durata bolii cu debut bulbar este în medie de 2,5 ani, iar cu debut spinal - 3,5 ani.
Doar 7% dintre pacienți trăiesc mai mult de 5 ani. Utilizarea riluzolului poate prelungi viața unui pacient cu o medie de 3 luni. Durata bolii este mai scurtă odată cu debutul bulbar al ALS (paralizia bulbară progresivă), cu o vârstă de debut mai mică de 45 de ani, precum și cu un tip de progresie rapidă pe scara ALS-FRS-R (pierderea mai multor peste 12 puncte pe an).
(ALS), cunoscută și sub numele de boala Lou Gehrig, este o boală degenerativă incurabilă a sistemului nervos central. Potrivit Statelor Asociației, doar jumătate dintre rezidenții din SUA au auzit de boală, aceeași imagine este observată în alte țări.
O modalitate de a atrage atenția la problema sclerozei laterale amiotrofice a devenit Ice Bucket Challenge, în care oamenii trebuie să-și toarne o găleată cu apă cu gheață și să facă o donație. În august 2014, campania a câștigat o popularitate deosebită în întreaga lume, atrăgând donații de 50 de milioane de dolari și peste 1,5 milioane de participanți. Președintele și CEO-ul 3 M Inge Thulin s-au alăturat numărului de participanți și a comentat despre participarea sa la acțiune:
"Scleroza laterala amiotrofica(ALS) este o boală teribilă. Am acceptat un apel de la familia angajatului nostru de peste 32 de ani, Allen Wahlgren, care suferă de această boală. A fost diagnosticat la începutul anului și acum este aproape complet paralizat. În urmă cu exact un an, l-am pierdut și pe unul dintre cei mai mari lideri ai afacerii stomatologice 3M, Larry Leer, care a murit din cauza SLA. Am văzut cât de repede a „ars”, a fost groaznic. Și am acceptat această provocare nu numai în onoarea lui Allen și Larry, ci și în onoarea tuturor familiilor care se confruntă cu această boală teribilă”.
Cauzele SLA
Cauza SLA este o mutație a anumitor proteine (ubiquitina) cu apariția unor agregate intracelulare. Formele familiale ale bolii sunt observate în 5% din cazuri. Practic, boala ALS afectează persoanele cu vârsta de peste patruzeci și șaizeci de ani, dintre care nu mai mult de 10% sunt purtători ai formei ereditare, oamenii de știință încă nu pot explica cazurile rămase prin influența oricăror influențe externe - ecologie, leziuni, boli și; alti factori.
Simptomele bolii
Simptomele precoce ale bolii includ convulsii, tresărire, amorțeală și slăbiciune la nivelul membrelor, precum și dificultăți de vorbire, dar astfel de semne se aplică unei game largi de boli. Acest lucru face diagnosticul foarte dificil până în perioada finală, când boala a intrat deja în stadiul de atrofie musculară.
Leziunile inițiale ale SLA pot apărea pe diferite părți ale corpului, până la 75% dintre pacienți boala debutează la nivelul extremităților, în principal cele inferioare.
Ce este? Cum se manifestă?
Manifestările inițiale ale bolii:
.slăbiciune în părțile distale ale brațelor, stângăciune la efectuarea mișcărilor fine ale degetelor, subțierea mâinilor și fasciculații (smușcări musculare)
.mai rar, boala debutează cu slăbiciune la nivelul brațelor proximale și a centurii scapulare, atrofie la nivelul mușchilor picioarelor în combinație cu parapareza spastică inferioară.
De asemenea, este posibil ca boala să înceapă cu tulburări bulbare - disartrie și disfagie (25% din cazuri)
Crampele (contracții dureroase, spasme musculare), adesea generalizate, apar la aproape toți pacienții cu SLA și sunt adesea primul semn al bolii
Manifestări clinice caracteristice SLA
Scleroza laterală amiotrofică se caracterizează prin afectarea combinată a neuronului motor inferior (periferic) și afectarea neuronului motor superior (tractul piramidal și/sau celulele piramidale ale cortexului motor al creierului).
Semne de afectare a neuronului motor inferior:
- slăbiciune musculară (pareză)
- hiporeflexie (reflexe scăzute)
- atrofie musculară
- fasciculații (contracții spontane, rapide, neregulate ale fasciculelor de fibre musculare)
Semne de deteriorare a neuronului motor superior:
- slăbiciune musculară (pareză).
- spasticitate (creșterea tonusului muscular)
- hiperreflexie (reflexe crescute)
- semne patologice ale piciorului și mâinii
Pentru ALS în majoritatea cazurilor caracterizat prin asimetria simptomelor.
În muşchii atrofiaţi sau chiar aparent intacţi se găsesc fasciculatii(smulsuri musculare), care pot apărea într-un grup muscular local sau pot fi răspândite.
Într-un caz tipic, debutul bolii este cu pierderea mușchilor tenari una dintre mâini cu dezvoltarea slăbiciunii aducției (aducției) și opoziției degetului mare, (de obicei asimetric), ceea ce face dificilă prinderea cu degetul mare și arătător și duce la tulburări în controlul motorului fin în mușchii degetului. mână. Pacientul întâmpină dificultăți în a ridica obiecte mici, a fixa nasturi și a scrie.
Apoi, pe măsură ce boala progresează, mușchii antebrațului sunt implicați în proces, iar mâna capătă aspectul unei „labe cu gheare”. Câteva luni mai târziu, o leziune similară se dezvoltă pe celălalt braț. Atrofia, răspândită treptat, afectează mușchii umărului și ai centurii scapulare.
În același timp sau mai târziu Adesea se dezvoltă leziuni ale mușchilor bulbari: fasciculații și atrofie a limbii, pareza palatului moale, atrofie a mușchilor laringelui și faringelui, care se manifestă sub formă de disartrie (tulburări de vorbire), disfagie (tulburări de deglutiție), și saliva.
Mușchii expresiei faciale și ai masticației sunt de obicei afectați mai târziu decât alte grupe musculare. Pe măsură ce boala progresează, devine imposibil să scoateți limba, să umflați obrajii sau să trageți buzele într-un tub.
Uneori se dezvoltă slăbiciune a extensorilor capului, din cauza căreia pacientul nu își poate ține capul drept.
Când diafragma este implicată în proces Se observă o respirație paradoxală (stomacul se scufundă când inspiră și iese în afară când expiră).
Picioarele sunt de obicei primele care se atrofiază grupele musculare anterioare și laterale, care se manifestă printr-un „picior în cădere” și un mers de tip steppage (pacientul își ridică piciorul sus și îl aruncă înainte, coborându-l brusc).
Este caracteristic că atrofia musculară este selectivă.
- Se observă atrofie pe mâini:
tenara
ipotenar
muşchii interosoşi
muschii deltoizi
- Pe picioare sunt implicați mușchii care efectuează flexia dorsală a piciorului.
- În mușchii bulbari sunt afectați mușchii limbii și ai palatului moale.
Sindromul piramidei se dezvoltă, de regulă, într-un stadiu incipient al SLA și se manifestă prin revigorarea reflexelor tendinoase. În urma acesteia, se dezvoltă adesea parapareza spastică inferioară. În mâini, reflexele crescute sunt combinate cu atrofia musculară, adică. se observă afectarea combinată, simultană, a căilor centrale (piramidale) și a neuronilor motori periferici, ceea ce este caracteristic SLA. Reflexele abdominale superficiale dispar pe măsură ce procesul progresează. Simptomul lui Babinski (atunci cand talpa piciorului este iritata cu dungi, degetul mare se extinde, celelalte degete de la picioare se extind si se extind) se observa in jumatate din cazurile de boala.
Pot exista tulburări senzoriale. La 10% dintre pacienți, se observă parestezii în părțile distale ale brațelor și picioarelor. Durerea, uneori severă, de obicei noaptea, poate fi asociată cu rigiditate articulară, imobilitate prelungită, spasme datorate spasticității mari, crampe (spasme musculare dureroase) și depresie. Pierderea sensibilității nu este tipică.
Tulburări oculomotorii nu sunt tipice și apar în stadiile terminale ale bolii.
Tulburările organelor pelvine nu sunt tipice, dar într-un stadiu avansat, pot apărea retenție urinară sau incontinență.
Deficiență cognitivă moderată(scăderea memoriei și a performanței mentale) apar la jumătate dintre pacienți. La 5% dintre pacienți se dezvoltă tipul frontal, care poate fi combinat cu sindromul parkinsonian.
O caracteristică a SLA este absența escarelor chiar și la pacienții paralizați imobilizați la pat.
Oriunde apar primele semne de SLA, slăbiciunea musculară este transferată treptat în tot mai multe părți ale corpului, deși cu forma bulbară a SLA, pacienții pot să nu trăiască până să vadă pareza completă a membrelor din cauza stopului respirator.
În timp, pacientul își pierde capacitatea de a se mișca independent. boala ALS nu afectează dezvoltarea mentală, cu toate acestea, cel mai adesea, începe depresia profundă - persoana așteaptă moartea. În stadiile finale ale bolii sunt afectați și mușchii care îndeplinesc funcția respiratorie, iar viața pacienților trebuie susținută de ventilație artificială și alimentație artificială. Durează 3-5 ani de la observarea primelor semne de SLA până la moarte. Cu toate acestea, există cazuri larg cunoscute în care starea pacienților cu boală ALS clar recunoscută s-a stabilizat în timp.
CINE ARE BAS?
Există peste 350.000 de pacienți cu SLA în întreaga lume.
În fiecare an, la 100.000 de locuitori, 5-7 persoane sunt diagnosticate cu SLA. Peste 5.600 de americani sunt diagnosticați cu SLA în fiecare an. Adică 15 cazuri noi de Bass pe zi
ALS poate afecta pe oricine. Rata de incidență (număr de noi) a SLA - 100.000 de persoane pe an
Mai puțin de 10% din cazurile de SLA sunt ereditare. SLA poate afecta atât bărbații, cât și femeile. SLA afectează toate grupurile etnice și socioeconomice
ALS poate afecta adulții tineri sau foarte în vârstă, dar este cel mai adesea diagnosticată la vârsta adultă mijlocie și târzie.
Persoanele cu SLA au nevoie de echipamente scumpe, tratament și îngrijire constantă 24 de ore din 24
90% din sarcina îngrijirii cade pe umerii membrilor familiei persoanelor cu SLA. SLA duce la o posibilă epuizare a resurselor fizice, emoționale și financiare. În Rusia, există peste 8.500 de persoane cu SLA la Moscova, există peste 600 de persoane cu SLA, deși acest număr este subestimat. Cei mai cunoscuți ruși care s-au îmbolnăvit de SLA sunt Dmitri Şostakovici, Vladimir Migulya.
Cauzele bolii sunt necunoscute. Nu există tratament pentru ALS. A existat o încetinire a progresiei bolii. Extinderea vieții este posibilă cu ajutorul unui ventilator de acasă.
Sindroamele care nu se pot distinge clinic de SLA clasică pot rezulta din:
Leziuni structurale:
tumori parasagitale
tumori ale foramenului magnum
spondiloza coloanei cervicale
sindromul Arnold-Chiari
hidromielie
anomalie arteriovenoasă a măduvei spinării
Infecții:
bacteriene - tetanos, boala Lyme
virale - poliomielita, herpes zoster
mielopatie retrovirală
Intoxicații, agenți fizici:
toxine - plumb, aluminiu, alte metale.
medicamente - stricnină, fenitoină
soc electric
radiații cu raze X
Mecanisme imunologice:
discrazie cu celule plasmatice
poliradiculoneuropatia autoimună
Procese paraneoplazice:
paracarcinomatoasă
paralimfomatos
Tulburări metabolice:
hipoglicemie
hiperparatiroidism
tireotoxicoza
deficit de acid folic,
vitaminele B12, E
malabsorbție
Tulburări biochimice ereditare:
defect al receptorului de androgeni - boala Kennedy
deficit de hexosaminidază
deficit de a-glucozidază - boala Pompe
hiperlipidemie
hiperglicinurie
metilcrotonilglicinurie
Toate aceste afecțiuni pot provoca simptomele întâlnite în SLA și ar trebui luate în considerare în diagnosticul diferențial.
Nu există un tratament eficient pentru boală. Singurul medicament, inhibitorul de eliberare a glutamatului riluzol (Rilutek), întârzie moartea cu 2 până la 4 luni. Se prescrie 50 mg de două ori pe zi.
Tratamentul bolii ALS
Baza tratamentului este terapia simptomatică:
- Fizioterapie.
Activitate fizica. Pacientul ar trebui să mențină activitatea fizică cât mai bine pe măsură ce boala progresează, apare nevoia unui scaun cu rotile și a altor dispozitive speciale.
.Cura de slabire. Disfagia creează un risc de intrare a alimentelor în tractul respirator. Uneori este nevoie de o sondă de alimentare sau de gastrostomie.
- Utilizarea dispozitivelor ortopedice: guler cervical, diverse atele, dispozitive pentru apucarea obiectelor.
Pentru crampe (spasme musculare dureroase): sulfat de chinină 200 mg de două ori pe zi sau fenitoină (Difenin) 200-300 mg/zi sau carbamazepină (Finlepsin, Tegretol) 200-400 mg/zi și/sau vitamina E 400 mg de două ori pe zi, precum și preparate cu magneziu, verapamil (Isoptin).
Pentru spasticitate: baclofen (Baclosan) 10 - 80 mg/zi, sau tizanidină (Sirdalud) 6 - 24 mg/zi, precum și clonazepam 1 - 4 mg/zi, sau memantină 10 - 60 mg/zi.
- Pentru a saliva, atropina 0,25 - 0,75 mg de trei ori pe zi, sau hioscina (Buscopan) 10 mg de trei ori pe zi.
Dacă este imposibil să mănânci din cauza tulburării de deglutiție, se pune o sondă de gastrostomie sau se introduce o sondă nazogastrică. Gastrostomia endoscopică percutanată precoce prelungește viața pacienților cu o medie de 6 luni.
- Pentru sindroamele dureroase se folosește întregul arsenal de analgezice. Inclusiv analgezice narcotice în etapele finale.
- Uneori, medicamentele anticolinesterazice (neostigmină metil sulfat - prozerină) aduc unele îmbunătățiri temporare.
Cerebrolizină în doze mari (10-30 ml picături intravenoase timp de 10 zile în cure repetate). Există o serie de studii mici care arată eficacitatea neuroprotectoare a Cerebrolysin în SLA.
.Antidepresive: Sertaline 50 mg/zi sau Paxil 20 mg/zi sau Amitriptiline 75-150 mg/zi (medicamentul este mai ieftin, dar are efecte secundare mai pronunțate; unii bolnavi de SLA îl preferă tocmai din cauza efectelor secundare - provoacă uscăciune în gură, reduce în consecință hipersalivația (salivația), care afectează adesea pacienții cu SLA).
.Dacă apar tulburări respiratorii: ventilația artificială în spitale, de regulă, nu se realizează, dar unii pacienți achiziționează ventilatoare portabile și efectuează ventilație mecanică la domiciliu.
- Sunt în curs de dezvoltare pentru utilizarea hormonului de creștere și a factorilor neurotrofici în SLA.
- Recent, a existat o dezvoltare activă a tratamentului cu celule stem. Această metodă promite a fi promițătoare, dar este încă în stadiul de experimente științifice.
Medicamente, medicamente care ajută la încetinirea bolii ALS:
Scleroza laterală amiotrofică (ALS, boala Gehrig, boala neuronului motor) a fost descrisă pentru prima dată de psihiatrul francez Martin Charcot în 1869.
În SUA și Canada, un alt termen este „boala lui Lou Gehrig”, un celebru jucător de baseball care a trebuit să-și încheie cariera la vârsta de 36 de ani din cauza sclerozei laterale amiotrofice.
Ce este scleroza laterală amiotrofică?
Scleroza laterală amiotrofică este o boală a sistemului nervos care afectează rapid neuronii motori din măduva spinării, trunchiul cerebral și cortexul.
Procesul patologic implică nervii motori ai neuronilor cranieni (faciali, ternari, glosofaringieni).
Scleroza laterală amiotrofică este rară (2-3 persoane la 100.000) și progresează rapid.
În medicină, există un alt concept - sindromul de scleroză laterală amiotrofică. Este provocată de o altă boală, astfel încât tratamentul în acest caz are ca scop eliminarea patologiei de bază. Dacă un pacient are simptome de SLA, dar cauzele acestora nu sunt cunoscute, medicii nu vorbesc despre un sindrom, ci despre o boală.
În ALS, neuronii motori sunt distruși, nu mai trimit semnale de la creier către mușchi, ca urmare, aceștia din urmă încep să slăbească și să se atrofieze.
Cauze

Motivele care provoacă apariția acestei boli nu au fost încă stabilite. Cu toate acestea, oamenii de știință oferă mai multe teorii:
Ereditar
S-a stabilit că în 10-15% din cazuri boala este ereditară.
Viral
Această teorie a devenit larg răspândită în anii 60 ai secolului XX în SUA și URSS. În acest moment, au fost efectuate experimente pe maimuțe. Animalelor li s-au injectat extracte din măduva spinării persoanelor bolnave. De asemenea, sa presupus că boala ar putea fi cauzată de virusul poliomielitei.
Gennaya
Dereglarea genelor se găsește la 20% dintre pacienții cu SLA. Ei codifică enzima Superoxid dismutaza-1, care transformă Superoxidul, care este periculos pentru celulele nervoase, în oxigen.
Autoimună
Oamenii de știință au efectuat cercetări și au descoperit anticorpi care ucid celulele nervoase motorii. S-a dovedit că acești anticorpi se pot forma în boli grave (limfom Hodgkin, cancer pulmonar etc.).
Neurale
Această teorie a fost dezvoltată de oamenii de știință britanici care cred că formarea SLA poate fi provocată de elemente gliale - celule responsabile de funcționarea neuronilor. Dacă funcția astrocitelor, care elimină glutamatul din terminațiile nervoase, este afectată, riscul bolii Charcot crește de câteva ori.
Printre factorii de risc, medicii identifică: predispoziția ereditară, vârsta peste 50 de ani, fumatul, munca care implică utilizarea plumbului și serviciul militar.
Simptomele ALS
Indiferent de forma bolii, toți pacienții simt slăbiciune musculară, apar zvâcniri musculare, iar masa musculară scade.
Slăbirea musculară crește rapid, dar mușchii ochiului și sfincterul vezicii urinare nu sunt afectați.
Într-un stadiu incipient al bolii, apar următoarele:
- slăbiciune musculară la glezne și picioare;
- atrofia mâinii;
- tulburări motorii și de vorbire;
- dificultate la inghitire;
- spasme musculare;
- crampe ale limbii, brațelor și umerilor.
Odată cu dezvoltarea SLA, apar atacuri de râs și plâns, echilibrul este perturbat și apare atrofia limbii.
Funcțiile cognitive se deteriorează doar în 1-2% din cazurile de boală la alți pacienți, activitatea mentală nu se modifică.
În etapele ulterioare, pacientul dezvoltă depresie, începe să experimenteze întreruperi ale respirației și își pierde capacitatea de a se mișca independent.
Pacienții cu SLA încetează să mai fie interesați de cei dragi și de lumea exterioară, devin capricioși, neîngrădiți, labili din punct de vedere emoțional și agresivi. Când mușchii respiratori încetează să funcționeze, o persoană are nevoie de ventilație artificială.
Cursul bolii

Inițial, apar simptome care împiedică o persoană să ducă o viață plină: amorțeală musculară, crampe, zvâcniri, dificultăți de vorbire. Dar, de regulă, este dificil să se determine de la început cauza exactă a acestor tulburări.
În cele mai multe cazuri, ALS este diagnosticată în stadiul de atrofie musculară.
Treptat, slăbiciunea musculară se extinde și acoperă noi zone ale corpului, pacientul nu se poate mișca independent și încep problemele de respirație.
Pacienții cu SLA suferă rareori de demență, dar starea lor duce la depresie severă în așteptarea morții. În ultima etapă, o persoană nu mai poate mânca, merge sau respira singură, are nevoie de dispozitive medicale speciale.
Formele bolii
Formele bolii se disting prin localizarea mușchilor afectați.
Bulbarnaya
Sunt afectați nervii cranieni (9,10,12 perechi).
Pacienții cu forma bulbară a SLA încep să aibă probleme cu vorbirea, se plâng de dificultăți de pronunție și le este greu să-și miște limba.
Pe măsură ce boala progresează, actul de înghițire este întrerupt și alimentele se pot scurge prin nas. Într-un stadiu avansat al bolii, mușchii feței și gâtului se atrofiază complet, expresiile faciale dispar, iar pacienții cu SLA nu pot deschide gura sau mesteca alimente.
Cervicotoracic

Boala progresează în membrele superioare de ambele părți.
Inițial, disconfortul apare în mâini și devine dificil pentru o persoană să efectueze mișcări complexe cu mâinile, să scrie sau să cânte la instrumente muzicale. La examinare, medicul observă că mușchii brațului pacientului sunt încordați și reflexele tendinoase sunt crescute.
În stadiile ulterioare ale bolii, slăbiciunea musculară progresează și se extinde la antebrațe și umeri.
Lombosacral
![]()
Primul simptom este slăbiciune la nivelul extremităților inferioare.
Devine mai dificil pentru pacient să lucreze în picioare, să urce scările, să meargă cu bicicleta și să meargă pe distanțe lungi.
În timp, piciorul începe să se lase, mersul se schimbă, apoi mușchii picioarelor se atrofiază complet, persoana nu poate merge și se dezvoltă incontinența urinară și fecală.
Aproape 50% dintre pacienți suferă de forma cervicotoracică a SLA, 25% fiecare reprezintă formele lombo-sacrale și bulbare.
Diagnosticare
Neurologul folosește următoarele ca principale metode de diagnostic:
RMN al creierului și măduvei spinării
Folosind această metodă, este posibil să se detecteze degenerarea structurilor piramidale și atrofia părților motorii ale creierului.
Examene neurofiziologice
Pentru a detecta ALS, se folosesc TCMS (stimulare magnetică transcraniană), ENG (electroneurografie) și EMG (electromiografie).
Puncția cefalorahidiană
Se determină nivelul conținutului de proteine (normal sau crescut).
Teste biochimice de sânge
La pacienții cu SLA se observă o creștere a nivelului creatin fosfokinazei de 5 sau mai multe ori, acumularea de creatinine și uree și o creștere a AST și ALT.
Analiza genetică moleculară
Se studiază gena care codifică Superoxid Dismutaza-1.
Dar pentru a identifica diagnosticul, aceste metode nu sunt suficiente, în paralel se foloseste diagnosticul diferential pentru a confirma sau exclude boli:
- creier: encefalopatie discirculatorie, tumori ale fosei craniene posterioare, atrofie a sistemelor multiple.
- măduva spinării: tumori, siringomielie, leucemie limfocitară, amiotrofie spinală etc.
- muşchii: miozită, miodistrofie oculofaringiană, miotonie Rossolimo-Steiner-Kurshman.
- nervi periferici: neuropatie motorie multifocala, sindromul Personaj-Turner etc.
- legatura neuromusculara: sindromul Lambert-Eaton, miastenia gravis.
Tratament
Nu există un remediu pentru ALS, dar este posibil să încetiniți progresia acestei boli, să creșteți speranța de viață și să atenuați starea unei persoane.
În acest scop, se utilizează terapia complexă:
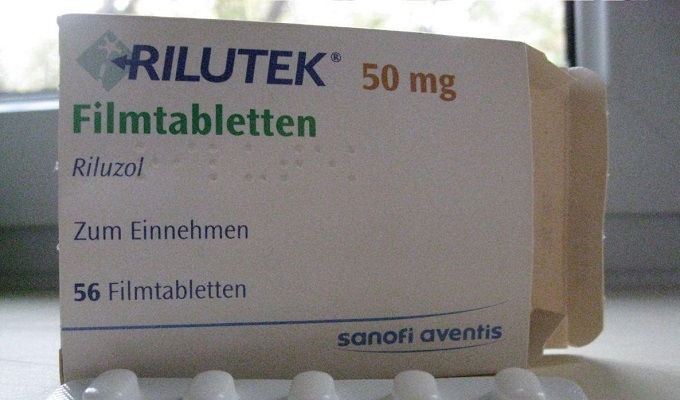
Un medicament care a fost utilizat pentru prima dată pentru tratamentul SLA în Marea Britanie și SUA. Substanțele active blochează eliberarea de glutamină și încetinesc procesul de afectare neuronală. Medicamentul trebuie luat de 2 ori pe zi, 0,05 g.
Relaxante musculare și antibiotice ajuta la rezolvarea slăbiciunii musculare. Pentru a elimina spasmele musculare și spasmele, sunt prescrise Mydocalm, Baclofen și Sirdalud.

Pentru a crește masa musculară, se folosește steroidul anabolic „Retabolin”.
Antibiotice dacă se dezvoltă sepsis sau apar complicații infecțioase. Medicii prescriu fluorochinolone, cefalosporine, carbopene.
Vitamine grupele B, E, A, C pentru a îmbunătăți impulsul de-a lungul fibrelor nervoase.
Medicamente anticolinesterazice, care încetinește procesul de distrugere a acetilcolinei („Kalimin”, „Prozerin”, „Pyridostigmină”).

În unele cazuri este folosit transplant de celule stem. Previne moartea celulelor nervoase, promovează creșterea fibrelor nervoase și restabilește conexiunile neuronale.
În etapele ulterioare este utilizat antidepresive și tranchilizante, analgezice nesteroidiene și opiacee.
Dacă somnul este perturbat, se prescriu medicamente benzodiazepine.
Pentru a facilita mișcările, sunt necesare scaune și paturi cu diverse funcții, bastoane și gulere de fixare. Medicii recomandă terapia logopedică. În stadiile ulterioare ale bolii, va fi nevoie de un ejector de salivă și apoi de o traheostomie pentru ca pacientul să poată respira.
Metodele de tratament netradiționale pentru SLA nu produc rezultate pozitive.
Prognoza si consecinte
Prognosticul pacienților cu SLA advers. Moartea survine după 2-12 ani, pe măsură ce se dezvoltă pneumonie severă, insuficiență respiratorie sau alte boli grave, cauzate de boala Gehrig.
În forma bulbară și la pacienții vârstnici, perioada se reduce la 3 ani.
Prevenirea
Măsurile de prevenire a SLA sunt încă necunoscute medicinii.
Boala progresează foarte repede, nu există cazuri de tratament cu succes și de restabilire a funcțiilor motorii ale organismului. Slăbiciunea musculară, care crește treptat, schimbă complet viața unei persoane și a familiei sale.
Dar, în ciuda prognozei dezamăgitoare și a studiului insuficient al bolii, cei dragi ar trebui să spere că în viitorul apropiat vor fi dezvoltate tratamente terapeutice eficiente. Între timp, este necesar să se ia măsuri pentru ameliorarea stării unei persoane cu SLA.
Scleroza laterală amiotrofică este o boală periculoasă care poate imobiliza o persoană și poate duce la moarte.
Medicii încă nu pot stabili cauzele exacte ale bolii și nu pot găsi tratamente eficiente. În momentul de față, tot ceea ce poate face medicina este să amelioreze starea pacienților cu SLA. Nici un singur pacient nu a reușit să se recupereze complet de această boală. Este important să distingem „boala ALS” de „sindromul ALS”. În al doilea caz, prognosticul pentru recuperare este mult mai bun.
Scleroza amiotrofică laterală (laterală). mai are numele boala ALS. Sunt cunoscute și alte denumiri pentru această boală: boala neuronului motor, boala Charcot, boala neuronului motor, boala Lou Gehrig. Ce este? Este o boală degenerativă incurabilă a sistemului nervos central care progresează lent, dar afectează atât cortexul cerebral, cât și nucleii măduvei spinării și nervilor cranieni. Acest lucru duce la deteriorarea neuronilor motori, paralizie și atrofie musculară.
Ca urmare, moartea apare din cauza insuficienței mușchilor respiratori sau din infecții ale tractului respirator. Poate fi observat și sindromul ALS, dar aceasta este o boală complet diferită.
Această boală a fost descrisă pentru prima dată de Charcot în 1869.
Cauzele SLA
Cauza SLA este o mutație a anumitor proteine (ubiquitina) cu apariția unor agregate intracelulare. Formele familiale ale bolii sunt observate în 5% din cazuri. Practic, boala ALS afectează persoanele cu vârsta de peste patruzeci și șaizeci de ani, dintre care nu mai mult de 10% sunt purtători ai formei ereditare, oamenii de știință încă nu pot explica cazurile rămase prin influența oricăror influențe externe - ecologie, leziuni, boli și; alti factori.
Simptomele bolii
Simptomele precoce ale bolii sunt amorțeală și slăbiciune la nivelul membrelor, precum și dificultatea de a vorbi, dar astfel de semne se aplică unui număr mare de boli. Acest lucru face diagnosticul foarte dificil până în perioada finală, când boala a intrat deja în stadiul de atrofie musculară.
Cel mai faimos pacient cu SLA: Stephen Hawking iubește găurile negre și televizorul
Leziunile inițiale ale SLA pot apărea pe diferite părți ale corpului, până la 75% dintre pacienți boala debutează la nivelul extremităților, în principal cele inferioare. Ce este? Există dificultăți de mers, pacientul începe să se poticnească și apare rigiditate în gleznă. Când extremitățile superioare sunt afectate, se pierde flexibilitatea degetelor și puterea mâinilor.
Se poate manifesta ALS (forma bulbară), care în etapa următoare progresează spre dificultăți la înghițire.
Oriunde apar primele semne de SLA, slăbiciunea musculară este transferată treptat în tot mai multe părți ale corpului, deși cu forma bulbară a SLA, pacienții pot să nu trăiască până să vadă pareza completă a membrelor din cauza stopului respirator.
În timp, pacientul își pierde capacitatea de a se mișca independent. boala ALS nu afectează dezvoltarea mentală, cu toate acestea, cel mai adesea, începe depresia profundă - persoana așteaptă moartea. În stadiile finale ale bolii sunt afectați și mușchii care îndeplinesc funcția respiratorie, iar viața pacienților trebuie susținută de ventilație artificială și alimentație artificială. Durează 3-5 ani de la observarea primelor semne de SLA până la moarte. Cu toate acestea, există cazuri larg cunoscute în care starea pacienților cu boală ALS clar recunoscută s-a stabilizat în timp.
CINE ARE BAS?
Există peste 350.000 de pacienți cu SLA în întreaga lume.
- pe an, 5-7 persoane la 100.000 de locuitori sunt diagnosticate cu SLA. Peste 5.600 de americani sunt diagnosticați cu SLA în fiecare an. Adică 15 cazuri noi de Bass pe zi
- ALS poate afecta pe oricine. Rata de incidență (număr de noi) a SLA - 100.000 de persoane pe an
- Mai puțin de 10% din cazurile de SLA sunt ereditare. SLA poate afecta atât bărbații, cât și femeile. SLA afectează toate grupurile etnice și socioeconomice.
- ALS poate afecta adulții tineri sau foarte în vârstă, dar este cel mai adesea diagnosticată la vârsta adultă mijlocie și târzie.
- Persoanele cu SLA au nevoie de echipamente scumpe, tratament și îngrijire constantă 24 de ore din 24
- 90% din sarcina îngrijirii cade pe umerii membrilor familiei persoanelor cu SLA. SLA duce la o posibilă epuizare a resurselor fizice, emoționale și financiare. În Rusia, există peste 8.500 de persoane cu SLA la Moscova, există peste 600 de persoane cu SLA, deși acest număr este subestimat. Cei mai cunoscuți ruși care s-au îmbolnăvit de SLA sunt Dmitri Şostakovici, Vladimir Migulya.
Cauzele bolii sunt necunoscute. Nu există tratament pentru ALS. A existat o încetinire a progresiei bolii. Extinderea vieții este posibilă cu ajutorul unui ventilator de acasă.



